Research presented at the 2015 American Epilepsy Society Annual Meeting in Philadelphia shows that early detection of cognitive difficulties using a brief computerized cognitive screening may help improve delays in intervention. Cognitive problems and behavioural problems in epilepsy are frequent.
Cognitive difficulties in children with epilepsy affect different domains, including memory, language, and executive function that occur during critical periods of development. However screening tools currently available do not adequately identify these children, resulting in a delay in intervention.
In their study Megan E. Bone, of the University of Pittsburgh School of Medicine, and colleagues evaluated the feasibility of the brief CNS Vital Signs (CNSVS) computerized cognitive battery for the detection of comorbidities in children with new-onset epilepsy.
They studied 33 patients (17 male; 26 generalized epilepsy, 7 focal epilepsy) aged 8-17 years with new-onset epilepsy and no previous antiepileptic drug treatment. Patients completed the CNSVS, which is completed in approximately 30 minutes, and parents completed the Strength and Difficulties Questionnaire, at two subsequent intervals (2-12 months = T2, 12-18 months= T3) during routine clinical appointments. Baseline scores were compared to subsequent scores using the Reliable Change Index (RCI).
All participants completed at least one follow up testing (mean follow-up time = 5.5 mo.), and 16 completed a third testing (mean total follow-up time = 14.5 mo.). Between baseline and first follow-up, 85% of patients had clinically significant changes in one of more cognitive domains including memory, psychomotor speed, reaction time, complex attention, and cognitive flexibility.
There was no apparent relationship between change in cognitive performance and seizure medication, epilepsy type, and seizure control. Composite memory, cognitive flexibility, reaction time, and complex attention showed the most change at both follow-up intervals, while no significant changes were observed for psychomotor speed.
In patients who improved or declined, change occurred at the first follow-up and remained stable thereafter. Parental concerns did not necessarily align with CNSVS score changes.
Citation: Bone ME, Triplett R, Rubin P, Asato MR. Abstract 1.293. Brief computerized screening detects cognitive changes in children with epilepsy. Presented at: American Epilepsy Society Annual Meeting; Dec 4-8, 2015; Philadelphia.
CNS Vital Signs (CNSVS) computerized cognitive battery
Read More
- Details
- ICNA
- News
- Hits: 870
Although novel treatment strategies based on the gene therapy approach for epilepsy has been encouraging, there is still a gap in demonstrating a proof-of-concept in a clinically relevant animal model and study design.
In a study published in Neurobiology of Disease on Dec 1, 2015 researchers from Lund University in Sweden and colleagues at University of Copenhagen Denmark delivered genes for a signal substance "neuropeptide Y" and its receptor into the brain of test animals with post intrahippocampal kainate-induced status epilepticus (SE) model of chronic epilepsy, and succeeded in considerably reducing the number of epileptic seizures among the animals. The test has been designed to as far as possible mimic a future situation involving treatment of human patients.
Neuropeptide Y (NPY), which is widely expressed in the brain, is involved in various brain functions, including regulation of neuronal excitability and seizures.
The intrahippocampal kainate model resembles the disease development of human chronic mesial temporal lobe epilepsy (mTLE) in that spontaneous seizures originate in the sclerotic hippocampus, only a part of the animals develops chronic epilepsy, animals show largely variable seizure frequency and tends to progressively increase over time.
Despite significant hippocampal degeneration caused by the kainate injection, the use of MRI allowed targeting the recombinant adeno-associated viral (rAAV) vectors encoding NPY and Y2 receptor genes to the remaining dorsal and ventral hippocampal areas ipsilateral to the kainate injection.
The study results have so far been positive.Continuous video-EEG monitoring demonstrated not only prevention of the progressive increase in seizure frequency in rAAV-NPY/Y2 treated animals as compared to the controls, and for 80 % of the animals the number of seizures was reduced by almost half.
This translationally designed study in a clinically relevant model of epilepsy suggests that simultaneous overexpression of NPY and Y2 receptors unilaterally in the seizure focus is a relevant and promising approach that can be further validated in more extensive preclinical studies.
Gene therapy treatments could initially be carried out on patients who have already been selected for surgical procedures. In the long term, however, gene therapy will be of the greatest benefit to those patients who cannot be operated on.
There are patients with severe epilepsy whose epileptic focus is so badly placed that an operation is out of question since it can impair e.g. speech or movement. These patients can therefore never undergo a surgical procedure, but could be helped by gene therapy in the future.
Citation: Translational approach for gene therapy in epilepsy: Model system and unilateral overexpression of neuropeptide Y and Y2 receptors, Ledri LN, Melin E, Christiansen SH, Gøtzsche CR, Cifra A, Woldbye DP, Kokaia M, Neurobiology of Disease , doi: 10.1016/j.nbd.2015.11.014, published online 1 December 2015.
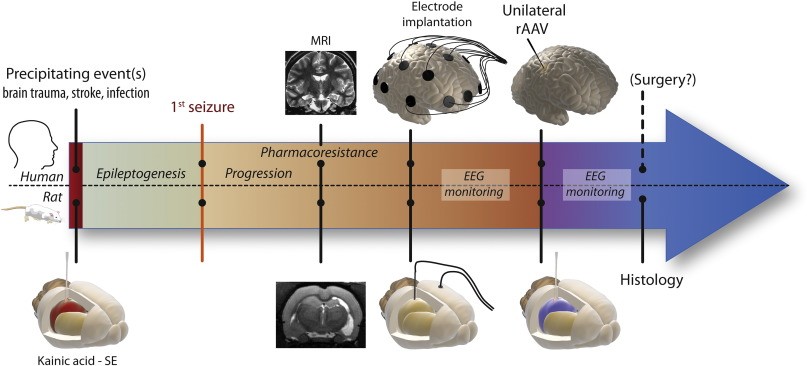
Cover image: Translational pre-clinical design of the study. A schematic overview of plausible interventions for epilepsy treatment using viral vector-based gene therapy for epilepsy patients. The pre-clinical study in rats has been designed based on a clinical perspective for possible future clinical trials in patients with intractable temporal lobe epilepsy and that are candidates for surgical resection of the epileptic focus.
Read More
- Details
- ICNA
- News
- Hits: 685
In a study, published Dec. 4, 2015 in the journal Science, investigators from the Bench to Bassinet Program’s Pediatric Cardiac Genomics Consortium used exome sequencing to genetically evaluate 1,213 family trios composed of a child with congenital heart disease and the mother and father. Through this technique, which examines only the protein-coding regions of DNA, they found that children with moderate-to-severe congenital heart disease had a substantial number of "de novo" gene mutations. De novo mutations occur within egg, sperm, and fertilized cells, but are not part of the genetic makeup of the mother or father.
Congenital heart disease (CHD) patients have an increased prevalence of extracardiac congenital anomalies (CAs) and risk of neurodevelopmental disabilities (NDDs). Exome sequencing of 1213 CHD parent-offspring trios identified an excess of protein-damaging de novo mutations, especially in genes highly expressed in the developing heart and brain.
These mutations accounted for 20% of patients with Congenital heart disease (CHD), neurodevelopmental disabilities (NDDs), and extracardiac congenital anomalies (CAs) but only 2% of patients with isolated CHD. Mutations altered genes involved in morphogenesis, chromatin modification, and transcriptional regulation, including multiple mutations in RBFOX2, a regulator of mRNA splicing. Genes mutated in other cohorts examined for NDD were enriched in CHD cases, particularly those with coexisting NDD. These findings reveal shared genetic contributions to CHD, NDD, and CA and provide opportunities for improved prognostic assessment and early therapeutic intervention in CHD patients.
These findings have implications for basic research and clinical medicine. Further analyses of these mutated genes, would help uncover new pathways that are critical for the development of the heart, brain, and other organs.
If the relationship between the de novo mutations and neurodevelopmental abnormalities in children continues to hold, clinical genetic tests could be created for newborns with moderate-to- severe congenital heart abnormalities. The patients found to carry the gene mutations could then be targeted for greater surveillance and early interventions that might address and limit developmental delays and improve their outcomes.
Congenital heart disease in which there are structural defects in the heart is the most common type of birth defect in the United States, and one of the leading causes of infant death. Nearly 40,000 children are born with congenital heart disease each year, and experts estimate that approximately 1 to 2 million adults and 800,000 children in the U.S. currently live with the disease.Surgery is often performed early in life to repair heart defects,but once children reach school age, many exhibit various attention deficits, including attention deficit hyperactivity disorder, and other neurobehavioral problems.
Citation:
Bench to Bassinet Program’s Pediatric Cardiac Genomics Consortium
Read More
- Details
- ICNA
- News
- Hits: 690

The medium chain triglyceride(MCT) ketogenic diet is an established treatment for drug-resistant epilepsy that increases plasma levels of decanoic acid and ketones. The general assumption is that the diet’s antiepileptic effect is due to ketone production.However there is a poor correlation between serum ketones and seizure control.
Recently decanoic acid within the MCT ketogenic diet has been shown to block seizures among people with epilepsy to a greater extent than medications currently used to treat the condition. Besides decanoic acid may even have fewer side effects.Indeed, in vitro, decanoic acid is more potent than valproic acid [a branched chain fatty acid isomer of octanoic acid],a common antiepileptic drug.
However the therapeutic mechanism of the MCT ketogenic diet remains unclear. In research published online in Brain, a team of researchers at the Royal Holloway University of London and University College London, has shown that a major constituent of the MCT ketogenic diet, decanoic acid, but not the ketones β-hydroxybutryate or acetone, exhibits antiseizure activity in two acute ex vivo rat hippocampal slice models of epileptiform activity.
Using heterologous expression of excitatory ionotropic glutamate receptor AMPA subunits in Xenopus oocytes, they show that this effect is through direct AMPA receptor inhibition, a target shared by a recently introduced epilepsy treatment perampanel.
Decanoic acid acts as a non-competitive antagonist at therapeutically relevant concentrations, in a voltage- and subunit-dependent manner, and this is sufficient to explain its antiseizure effects. This inhibitory effect is likely to be caused by binding to sites on the M3 helix of the AMPA-GluA2 transmembrane domain; independent from the binding site of perampanel.
These results indicate that the direct inhibition of excitatory neurotransmission by decanoic acid in the brain contributes to the anti-convulsant effect of the medium chain triglyceride ketogenic diet. According to Prof Robin Williams at Royal Holloway University, the possibility that mechanism of action of the ketogenic diet is related to the fat in the diet rather than the ketones will lead to the development of improved diets and suggests that the ketogenic diet should be renamed simply as the "MCT diet".
It is important to note, however, that there may be circumstances where ketones do play a greater role in seizure control such as in Glut1 deficiency, the ketones from the ketogenic diet providing the brain with an alternative energy source. In addition the ketogenic diet in the longer term may modify metabolic and gene expression, which could have important disease-modifying effects.
Citation
Chang P, Augustin K, Boddum K, Williams S, Sun M, Terschak JA, Hardege JD, Chen PE, Walker MC, Williams RS (2015) Seizure control by decanoic acid through direct AMPA receptor inhibition. BRAIN 2015. DOI: http://dx.doi.org/10.1093/brain/awv325 First published online: 25 November 2015
cover photo: courtesy The Charlie Foundation for ketogenic therapies
Read More
- Details
- ICNA
- News
- Hits: 778

SPRITAM (levetiracetam) has been approved in the form of a dissolvable pill for the treatment of seizures among people with epilepsy.
Each year, around 150,000 people in the US are diagnosed with epilepsy. Of the 2.9 million people living with the condition, around 460,000 are children.A 2002 study published in the journal Epilepsy and Behavior found that 71% of epilepsy patients reported forgetting, missing or skipping a dose of anti-seizure medication at some point. Almost half of these patients reported having a seizure after their missed dose.
The team at Aprecia Pharmaceuticals used their ZipDose Technology to develop SPRITAM - a technique that was originally created by researchers from the Massachusetts Institute of Technology (MIT).
This technology allows the drug to be layered and tightly packed into a single pill, which dissolves instantly with just one sip of liquid, making the medication much easier to administer.
Another advantage of the drug being created in this way is that up to 1,000 mg of the medication can be loaded into one dose. In addition, the layering technology allows drug doses to be closely controlled and tailored to the need of each individual patient.
It is hoped that the Food and Drug Administration's (FDA) approval of SPRITAM will improve medication adherence among epilepsy patients, and its creators believe it may open the door to the development of new drugs for the treatment of other neurological disorders. Aprecia say SPRITAM will be available from early next year.
Read More
- Details
- ICNA
- News
- Hits: 1004