 Preliminary results from the FEBSTAT study has revealed that within days of a prolonged fever-related seizure, some children have signs of acute brain injury, abnormal brain anatomy, altered brain activity, or a combination.Febrile Seizures affect 3 to 4 percent of all children. Most such children recover rapidly and do not suffer long-term health consequences. However, having one or more prolonged febrile seizures in childhood is known to increase the risk of subsequent epilepsy which is estimated to be about 30-40 percent following febrile status epilepticus (FSE).
Preliminary results from the FEBSTAT study has revealed that within days of a prolonged fever-related seizure, some children have signs of acute brain injury, abnormal brain anatomy, altered brain activity, or a combination.Febrile Seizures affect 3 to 4 percent of all children. Most such children recover rapidly and do not suffer long-term health consequences. However, having one or more prolonged febrile seizures in childhood is known to increase the risk of subsequent epilepsy which is estimated to be about 30-40 percent following febrile status epilepticus (FSE).
The Consequences of Prolonged Febrile Seizures in Childhood (FEBSTAT) study is focused specifically on FSE and the risk of temporal lobe epilepsy.Within days of FSE, the children in the study underwent magnetic resonance imaging (MRI) and electroencephalography (EEG). The MRI findings were reported in July 2012, and the EEG findings were reported in November 2012. Both papers were published in Neurology.
FEBSTAT began almost 10 years ago. 199 Children, aged between one month and six years with FSE were enrolled from 2003-2010 at five sites: Montefiore Medical Center and Jacobi Hospital in the New York City; Lurie Children's Hospital in Chicago; Duke University Medical Center in Durham, N.C.; Virginia Commonwealth University Hospital in Richmond; and Eastern Virginia Medical School in Norfolk.A further 96 children who had experienced a single, simple febrile seizure (lasting 10 minutes or less) were also recruited and served as the comparison (control) group.
The children underwent blood tests, a neurological exam, MRI, and EEG within 72 hours of being seen in the emergency room for FSE. The blood samples are being analyzed for viral infections linked to febrile seizures and for the presence of gene mutations that could contribute to epilepsy. The children continue to receive neurological exams, MRI and EEG at regular intervals.
The MRI scans revealed that FSE is sometimes associated with abnormalities in the hippocampus. Of 191 children with FSE, 22 (11.5 percent) had signs of hippocampal injury on MRI, and 20 (10.5 percent) had developmental abnormalities of the hippocampus. Abnormal MRI results were rare among children with simple febrile seizures, defined as lasting 10 minutes or less. Out of 96 children with such seizures, only two (2.1 percent) had developmental abnormalities of the hippocampus and none had signs of brain injury.
Nearly half (45.2 percent) of the children with FSE had abnormal EEG findings. There was also a correlation between the MRI and EEG findings. Children with evidence of acute brain injury after FSE were more than twice as likely to have abnormal EEG findings. The results suggested that in some children prolonged febrile seizures do result in brain injury. In some others pre-existing brain abnormalities may increase the susceptibility to febrile seizures. Both these situations could eventually lead to epilepsy. But it will take more time for the study to confirm this, especially as it usually takes between 8 and 11 years for TLE to develop following FSE.
Temporal lobe epilepsy can cause memory loss, and neuroimaging of adults with temporal lobe epilepsy have sometimes revealed shrinkage and cell loss within the temporal lobe and hippocampus. Many adults with the disorder also report a history of FSE.
If MRI and EEG findings associated with FSE ultimately do correlate with epilepsy, they could be used to identify kids who are at risk and who might benefit from research on preventative therapies for epilepsy.
FEBSTAT is supported by grant NS043209 to Dr. Shinnar. Data from children with simple febrile seizures were taken from a study led by Dale C. Hesdorffer, Ph.D., of Columbia University in New York. That study ended in 2004, and was supported by grant HD036867 from NIH's Eunice Kennedy Shriver National Institute of Child Health and Human Development.

References:
- M. J. Berg, B. Abou-Khalil. Childhood febrile status epilepticus: Chicken or egg? Does it matter? Neurology, 2012; 79 (9): 840 DOI: 10.1212/WNL.0b013e3182684559
- S. Shinnar, J. A. Bello, S. Chan, D. C. Hesdorffer, D. V. Lewis, J. MacFall, J. M. Pellock, D. R. Nordli, L. M. Frank, S. L. Moshe, W. Gomes, R. C. Shinnar, S. Sun. MRI abnormalities following febrile status epilepticus in children: The FEBSTAT study. Neurology, 2012; 79 (9): 871 DOI: 10.1212/WNL.0b013e318266fcc5
Read More
- Details
- ICNA
- News
- Hits: 730
 Prof Dimitri Kullmann's team from University College London (UCL) writing in the November 2012 issue of Science Translational Medicine,reports the potential efficacy of gene therapy in focal neocortical epilepsy using a rodent model.
Prof Dimitri Kullmann's team from University College London (UCL) writing in the November 2012 issue of Science Translational Medicine,reports the potential efficacy of gene therapy in focal neocortical epilepsy using a rodent model.
The authors created a rat model of focal neocortical epilepsy where they induced epilepsy by injecting tetanus toxin into the rat motor cortex. In this model the animals show motor seizures and epileptic activity similar to Epilepsia Partialis Continua.
They then injected a lentiviral vector carrying the gene NpHR which encodes for the light-activated chloride ion pump halorhodopsin along with tetanus toxin. A miniaturized wireless system was used to record the EEG activity.A week later, through an optic fiber inserted into the epileptogenic region of the rat brain a laser light in 20-s pulses was delivered to activate halorhodopsin expressed by excitatory principal neurons .When the haloorhodopsin channels are activiated, chloride ions flow into the neurons resulting in membrane hyperpolarization, decreased neuronal excitability and epileptic activity. The controls rats who were not exposed to light did not show any activation of halorhodopsin no suppression of epileptic activity.
In another method using gene therapy they injected a lentiviral vector carrying the gene KCNA1 which encodes Kv1.1 the potassium ion channel.Kv1.1 expression results in an outward flow of potassoim ions, resulting in membrane hyperpolarisation and reduced neuronal excitability.In this situation there was no need for activation using an external light source. They found that following injection of the virus carryin the KCNA1 gene, the animals who have been injected with the tetanus toxin showed a steady reduction in epileptic actiivty. The epileptic activity eventually stopped completely with no resulting motor or behavioural deficits.
Gene therapy approaches has several advantages over drug treatment, in that their action is very specific by delivering the therapeutic gene to a small group of neurons in a specific brain region. Epileptogenic circuits in the brain often results in altered expression of multiple genes resulting in down-regulation of ion channels thereby making AEDs that work by targeting receptors or ion channels less effective.
This study is proof-of-concept that gene therapy can have disease-modifying effects in epilepsy. However this is still in a very early phase of research, and the challenge will be to translate these results into effective treatment for epilepsy.
The optogenetics approach,is disadvantageous in that it necessitates insertion of an optic fiber to deliver the laserlight which activates the injected gene. Besides there are safety issues in inducing the expression of a foreign protein like halorhodopsin. It is also not known for how long halorhodopsin delivered by gene therapy will be expressed by excitatory neurons and whether there are any side effects from long term expression of the halorhodopsin.
At present it is not certain whether viral transfection does not cause any long term risk . It is also not known whether the expression of Kv1.1 channels by principal neurons diminish over time.
This approach also needs to be validated in other animal models of epilepsy, such as temporal lobe epilepsy(TLE), since TLE invovles multiple epileptogenic regions and would require multiple injections of the viral vector. These advances are in the very early stages still and many questions remain unanswered, but certainly opens up exciting prospects for the future.
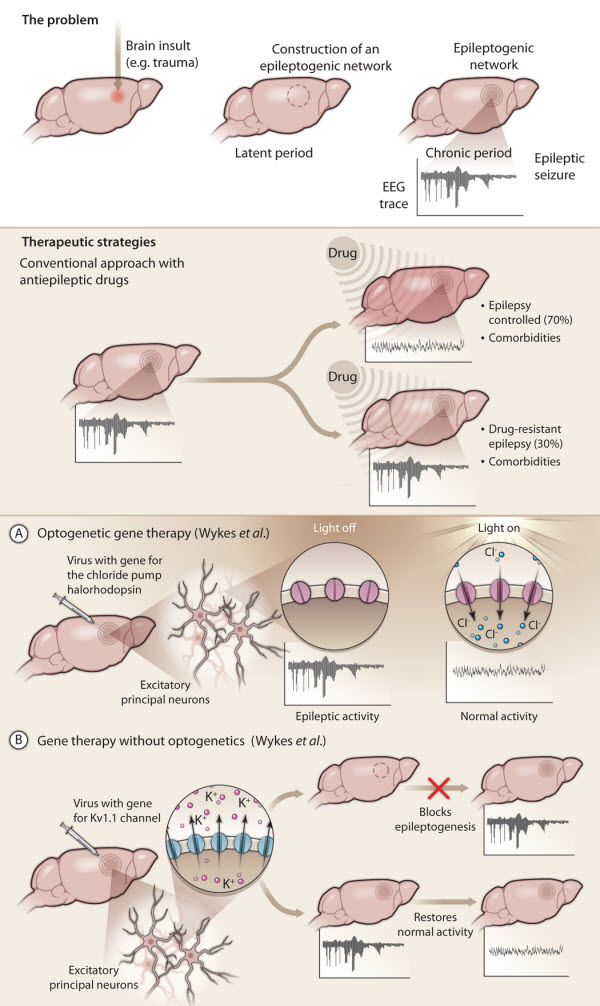
Fig. 1. Gene therapy for treating epilepsy.
(Top) Acquired epilepsies (in contrast to inherited epilepsies) may occur after an insult to the brain, such as trauma or meningitis. In response to the insult, specific neuronal networks in the brain become reorganized (hashed circle) in a process called epileptogenesis. Epileptic seizures are not yet apparent and this latent period can last for decades. At some point, the reorganized networks start to generate spontaneous seizures and full-blown epilepsy emerges (chronic period).
(Middle) Current antiepileptic drugs have many limitations. These drugs only control seizures in 70% of patients, with 30% of patients remaining refractory to drug treatment. None of the current antiepileptic drugs prevent the formation of epileptogenic networks and they often have severe side effects because they act nonspecifically in the brain.
(Bottom) Gene therapy for treating epilepsy specifically targets the affected brain region. In new work, Wykes et al.use two different gene therapy approaches to treat focal epilepsy induced in rat brain by injection of tetanus toxin into the motor cortex.
(A) Using an optogenetics approach, these investigators injected a viral vector carrying the gene for the light-sensitive chloride pump halorhodopsin (pink channels) together with tetanus toxin into rat brain. They used laser light delivered through an optic fiber to activate halorhodopsin expressed by excitatory principal neurons in the epileptogenic region. Activated halorhodopsin allowed chloride ions to flow into the neurons, membrane hyperpolarization, and a decrease in neuronal excitability leading to a reduction in epileptic activity.
(B) The second gene therapy approach involved injecting a viral vector carrying a gene encoding the native potassium ion channel Kv1.1 into rat brain. Overexpression of Kv1.1 (blue channels) by rat excitatory principal neurons resulted in an outward flow of potassium ions, increased membrane hyperpolarization, and decreased neuronal excitability. When virus with Kv1.1 was injected into rat brain at the same time as tetanus toxin, epileptogenesis was prevented.
When virus with Kv1.1 was injected into rat brain after epilepsy had been established, epileptic activity was progressively suppressed over several weeks, ultimately resulting in a normal EEG.
CREDIT: Y. HAMMOND/SCIENCE TRANSLATIONAL MEDICINE
Citations:
Wykes RC, Heeroma JH, Mantoan L, Zheng K, Macdonald DC, Deisseroth K et al. (2012) Optogenetic and Potassium Channel Gene Therapy in a Rodent Model of Focal Neocortical Epilepsy. Sci Transl Med ():. DOI: 10.1126/scitranslmed.3004190 PMID: 23147003.
Read More
- Details
- ICNA
- News
- Hits: 789
 In The Lancet, David Neal Franz and colleagues from Cincinnati Children’s Hospital Medical Center report on an international, double-blind, placebo-controlled phase 3 trial of the efficacy of the mTORC1 inhibitor everolimus in patients with subependymal giant cell astrocytomas associated with tuberous sclerosis complex. Everolimus is a rapamycin analogue with improved bioavailability compared with sirolimus.
In The Lancet, David Neal Franz and colleagues from Cincinnati Children’s Hospital Medical Center report on an international, double-blind, placebo-controlled phase 3 trial of the efficacy of the mTORC1 inhibitor everolimus in patients with subependymal giant cell astrocytomas associated with tuberous sclerosis complex. Everolimus is a rapamycin analogue with improved bioavailability compared with sirolimus.
This study is the latest to to show the effectiveness of everolimus in slowing the cell growth that is overactive in patients with TSC.
The phase III study was conducted among 117 patients with TSC who were randomly assigned to either everolimus or a placebo. Patients were 9 ½ years old on average but ranged from infants to adults. No patient on placebo showed improvement in their tumors. Tumor volume was measured by MRI assessment of the brain.Thirty-five percent of patients in this study on everolimus had at least a 50 percent reduction in tumor volume after an average of 42 weeks on medication.
Every patient in this study experienced a decrease in size of their tumors, and no patient required surgery for their tumors after treatment with everolimus. The earlier phase II study of everolimus by the same group was published in The New England Journal of Medicine in 2010. Based on that data, the U.S. Food and Drug Administration granted accelerated approval of everolimus for patients with subependymal giant cell astrocytomas, or SEGAs. The new, placebo-controlled study was conducted to confirm these earlier results.
In the current trial the patient reported quality of life, as measured by a validated quality of life and neuropsychological assessments, also improved at three months and six months after treatment with everolimus. The investigators also found that more patients had mutations in the TSC2 gene than in the TSC1 gene, and that TSC2 mutations correlated with lower response of the subependymal giant cell astrocytomas, which is in keeping with the results of earlier studies showing an increased severity of symptoms in patients with mutations in TSC2
Advances in the knowledge about tuberous sclerosis complex have shown that inactivating mutations in the Tsc1 and Tsc2 tumour suppressor genes leading to constitutive activation of the mammalian target of rapamycin (mTOR) as one of the main causes of astrocytomas.mTOR functions as an integrator of signalling within two multiprotein complexes (mTORC1 and mTORC2) leading to increased protein synthesis, and thus cell proliferation and survival. In the brain, growth of subependymal giant cell astrocytomas can cause life-threatening symptoms eg, hydrocephalus, requiring surgery. The same mTOR pathway associated with overactive cell growth in TSC also is implicated in other cancers and neurological conditions, such as Alzheimer’s disease, type 2 diabetes, Parkinson’s disease, Huntington’s disease and autism. This makes everolimus, an mTOR inhibitor, a potential candidate to treat these mTOR-associated disorders
Based on studies by John Bissler, MD, a nephrologist at Cincinnati Children’s, the US Food and Drug Administration (FDA) earlier this year approved everolimus as the first medication to shrink non-cancerous kidney tumors that strike up to 80 percent of people with TSC. This allows many patients to maintain kidney function for years to come without the need for repeated surgical intervention.Prior to FDA approval, surgery was considered standard therapy for SEGAs, but everolimus has now become a potential alternative to surgery and the first targeted medical therapy for TSC.
Citation: Franz DN, Belousova E, Sparagana S, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2012; published online Nov 14. (12)61134-9.[abstract]
Read More
- Details
- ICNA
- News
- Hits: 777
An evidence-based update to the 2004 guidelines for the treatment of infantile spasms has recently been published. Important new recommendations include use of low-dose adrenocorticotropic hormone (ACTH) over high-dose ACTH or vigabatrin. A paucity of data, however, leaves several key questions unanswered
- Details
- Raili Sylvia Riikonen
- News
- Hits: 4404