
In an article published in the Journal of Neuroscience Prof. John Chatham, of the Department of Pathology at the University of Alabama at Birmingham, and colleagues report that increasing O-GlcNAcylation levels in brain cells using the dietary supplement "glucosamine" widely used as a supplement to help reduce pain in osteoarthritis and other conditions was found to reduce reduce neural excitability in rodents.
The researchers had in a previous study shown that increases in protein O-GlcNAcylation are associated with a reduction in the strength of synapses in the hippocampus of the brain. This synaptic dampening effect on glutamatergic networks suggests that increasing O-GlcNAcylation will depress pathological hyperexcitability. Using in vitroand in vivo models of epileptiform activity, they have shown in the current study that acutely increasing O-GlcNAc levels can significantly attenuate ongoing epileptiform activity and prophylactically dampen subsequent seizure activity.
They tested this hypothesis using glucosamine which blocks an enzyme that clears O-GlcNAcylation from the brain, leading to a rapid increase in levels of the protein. Glucosamine was applied to hippocampal brain slices derived from rats and mice in which epileptiform activity was generated by GABAAR inhibition using drugs. The drug induced neural excitability was found to be dampened with ten minutes of the application of the compound glucosamine.
This is an exciting finding representing another target for research in epilepsy therapeutics.
Stewart LT, Khan AU, Wang K, Pizarro D, Pati S, Buckingham SC et al. (2017) Acute Increases in Protein O-GlcNAcylation Dampen Epileptiform Activity in Hippocampus.J Neurosci 37 (34):8207-8215. DOI: 10.1523/JNEUROSCI.0173-16.2017 PMID: 28760863.
Read More
- Details
- ICNA
- News
- Hits: 1562

In new research published in Neuron this week researchers from Brown University in Providence, RI suggests that disruption of the circadian rhythm protein CLOCK alters cortical circuits and may lead to generation of focal epilepsy.
Dr. Liu and her colleagues used resected brain tissue from epilepsy surgery for focal cortical dyplasias and analysed the tissue's transcriptome, or a survey of the messenger RNA (mRNA) in any given population of cells. They used adjacent healthy tissue as controls.The researchers were looking at differences between healthy and epileptogenic tissue and to their surprise found that there was a decrease in the expression of mRNA coding for a protein called circadian locomotor output cycles kaput (CLOCK) in the epileptogenic tissue.
CLOCK is a key player in the regulation of our circadian rhythms. Mice with mutated versions of the gene are unable to maintain normal daily rhythms of sleep and wakefulness. In the unhealthy brain tissue from participants with epilepsy, CLOCK was reduced in both excitatory and inhibitory neuron. CLOCK is important for regulating a range of genes and other CLOCK-associated proteins were also absent or reduced in the brain tissue.
To investigate this further the researchers generated and tested separate knock out mouse models with targeted deletions of the CLOCK gene in excitatory and inhibitory neurons. They found that the mice without CLOCK in their excitatory neurons showed symptoms of epilepsy similar to those in the human patients, including an increased susceptibility to seizures - especially when waking up.
They also found that excitatory neurons that lacked CLOCK received less inhibitory inputs from surrounding cells, in effect unleashing them and potentially giving them a lower threshold for the onset of seizures. The findings will certainly put more focus on the role of CLOCK in the molecular mechanisms behind epilepsy.
Citation: Li P, Fu X, Smith NA, Ziobro J, Curiel J, Tenga MJ et al. (2017) Loss of CLOCK Results in Dysfunction of Brain Circuits Underlying Focal Epilepsy.Neuron 96 (2):387-401.e6. DOI: 10.1016/j.neuron.2017.09.044 PMID: 29024662.
Read More
- Details
- ICNA
- News
- Hits: 1405
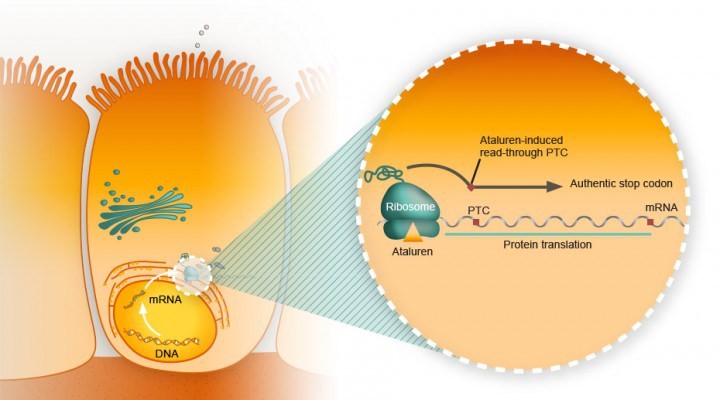
In a paper just published online in the journal The Lancet, Craig McDonald and colleagues at 53 study sites in 18 countries describe the clinical benefit of using the drug ataluren for a certain group of patients carrying a specific "nonsense mutation" for Duchenne muscular dystrophy.
Duchenne muscular dystrophy (DMD) is a progressive and life-limiting X-linked recessive disorder caused by mutations in the DMD gene that result in reduced or absent dystrophin production. Dystrophin is part of the dystrophin–glycoprotein complex, which acts as a scaffold between the actin cytoskeleton and the extracellular matrix and, as such, maintains muscle fibre integrity.
A single nucleotide change in the DNA sequence that introduces a premature stop codon is known as a nonsense mutation, a subset of a major class of premature termination codon (PTC) mutations. Nonsense mutations cause premature termination of translation resulting in the production of truncated polypeptides, which in turn halts the ribosomal translation process at an earlier site than normal, producing a truncated, non-functional protein. Ataluren makes ribosomes less sensitive to premature stop codons (referred to as "read-through"). Ataluren allows for those premature stop codons to be read through and complete the protein:
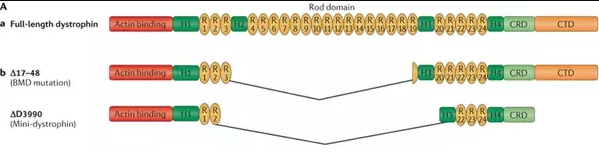
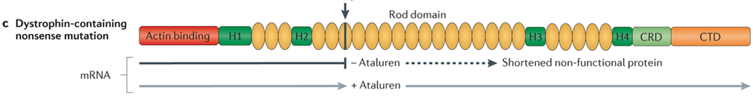
Ataluren has been marketed under the trade name Translarna by PTC Therapeutics (South Plainfield, NJ, USA). The drug allows ribosomal readthrough of premature stop codons, thus enabling production of functional dystrophin that might ameliorate disease progression.
The study, a phase 3, multicentre, randomised, double-blind, placebo-controlled trial (ACT DMD) was sponsored by the New Jersey-based company that developed ataluren, PTC Therapeutics. It included 230 patients, half of whom received the drug therapy over a course of 48 weeks, and the other half who received a placebo.
It assessed the ability of ataluren to stabilise ambulation, with a focus on a prespecified subgroup of patients with ambulatory decline. The primary endpoint of change in 6-min walk distance (6MWD) from baseline to week 48, with a hypothesis of a difference of at least 30 m between ataluren-treated and placebo-treated patients, did not differ significantly between groups in the intention-to-treat population.
Duchenne study participants with the nonsense mutation who had a baseline six-minute walk of between 300 meters and under 400 meters were the subgroup of patients who were the most likely to see the clinical benefits of ataluren.
ACT DMD and the use of ataluren provide a glimmer of hope for the approximately 10–15% of patients with DMD have an underlying nonsense mutation in the DMD gene 2 where the drug seems to prompt a slowing or stabilizing of the disease progression and motor function.
In 2016, the European Medicines Agency gave conditional approval for the drug to be used and, once data demonstrating its ability to stabilise ambulation were obtained, the National Institute for Health and Care Excellence (NICE) agreed reimbursement within a managed access agreement for the treatment of ambulant patients with DMD aged 5 years or older.
Other therapeutic approaches are currently being trialled in patients with DMD, several of which are mutation specific. These approaches include the use of compounds facilitating the upregulation of dystrophin analogues (NCT02858362); exon-skipping techniques with antisense oligonucleotides to convert an out-of-frame mutation into an in-frame mutation, thus allowing partial dystrophin expression; and the use of selective steroid receptor modulators (NCT02760264).
Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial, Craig M McDonald et al., The Lancet, doi: 10.1016/S0140-6736(17)31611-2, published 17 July 2017.
Wood CL, Cheetham T (2017) Treatment of Duchenne muscular dystrophy: first small steps.Lancet():. DOI: 10.1016/S0140-6736(17)31669-0 PMID: 28728957.
Read More
- Details
- ICNA
- News
- Hits: 1759

At the Amsterdam ICNC in May 2016 interested members of the ICNA met to discuss a way forward to provide support for trainees and newly qualified child neurologists (within 5 years)
Direction of how to learn and what to learn for trainees
Educational meetings – especially focused on-site educational meetings
How to put a grant together (Plan a symposium at the next ICNC2018 on this)
Support from the EB with guidance on projects – developing viable protocols
Big support for need for fellowships to support skills development in RPS
On-line training resources – identify and support ICNApedia as a resource and portal
Promotion of telemedicine to discuss complex cases
Guidance from established ICNA members on career development to trainees before taking up permanent posts and losing the capacity to adjust career path. Distance mentorship would avoid a conflict of interest but may need to include individuals from same country who know the local system best.
Learn how to advocate for patients and for what is needed for the profession (Plan a session at ICNC2018)
A working group need to be elected. This group would predominantly hold "virtual meetings", communicating via e-mail and skype and coming together at opportunistic meetings which they would be attending routinely e.g. ICNCs. The lead for this working group will be funded to attend the ICNA EB meetings (once a year) to feedback progress and developments in the program.
We call for nominations for members who would like to be part of this committee. There will be regional representation and the committee will rotate every 4 years. Two doctors from each of the ICNA regions (Africa,
Eastern Asia-Oceania, Europe, North America, South America, West Asia) will be elected to the committee. The person with the greatest number of votes will be the leader.
Please complete the online form if you would like to submit your nomination.
Read More
- Details
- ICNA
- News
- Hits: 1293
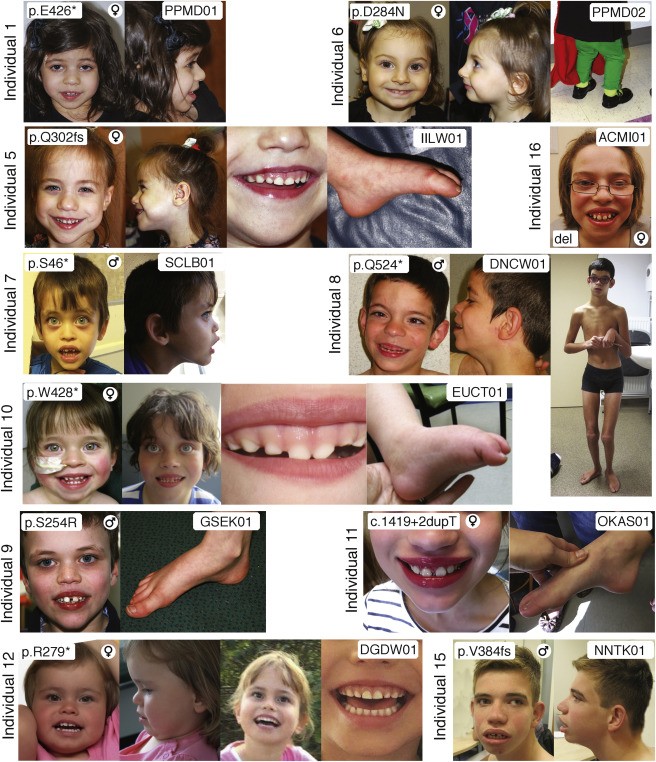
Researchers at Children's Hospital of Philadelphia (CHOP) have pinpointed variants in the WDR26 gene to a rare syndrome characterized by intellectual disability, seizures, an abnormal gait and distinctive facial features. In a study published in the American Journal of Human Genetics Deardorff, first author Cara M. Skraban, MD, also of CHOP, and co-authors from medical centers in six countries reported on 15 individuals now known to have this recognizable syndrome. Prior to this it was not even listed in some of the most commonly used databases.
The affected individuals ranged from two years old to 34 years old. All the patients had developmental delays (ranging from mild to severe), seizures, and similar facial features (such as wide mouths, prominent upper lip and gums, full cheeks and a broad nasal tip). Many had subtle abnormalities in their gait. All 15 had de novo (new) mutations - those arising in a single egg or sperm that developed into the affected patient, but did not occur in the patient's parents.
CHOP has started a patient registry to compile clinical data on this rare condition (see This email address is being protected from spambots. You need JavaScript enabled to view it.). The data collection may offer a resource for families interested in contacting each other to share information and support.
Source: WDR26 Haploinsufficiency Causes a Recognizable Syndrome of Intellectual Disability, Seizures, Abnormal Gait, and Distinctive Facial Features, Cara M. Skraban et al., American Journal of Human Genetics, doi: 10.1016/j.ajhg.2017.06.002, published 6 July 6 2017.
Read More
- Details
- ICNA
- News
- Hits: 1388